
De la Genómica a la Era de la Postgenómica: Craig Venter, un protagonista destacado.

Cuando en 1990 se inició el Proyecto del Genoma Humano (PGH) en Estados Unidos [1], nadie pudo anticipar el vertiginoso camino que iba a tomar el campo de la biología molecular y más concretamente la genética molecular. Desde entonces la ciencia biológica ha sufrido una transformación y una evolución sin precedentes. Se han sucedido los descubrimientos de mano de un desarrollo tecnológico impensable no solo en ese momento si no en los sucesivos años de esta carrera. Esto ha propiciado el nacimiento de las sucesivas ciencias “ómicas”, genómica, transcriptómica, metabolómica, proteóimica, entre otras, que han cambiado la forma que tenemos de analizar el mundo biológico a nivel molecular y por tanto el desarrollo consiguiente desarrollo científico. Durante este periodo han sido muchos los científicos que han contribuido a este desarrollo, pero quizás hay uno que es protagonista de excepción, Craig Venter. Este documento pretende presentar esta transición de la genética molecular a la llamada genómica y a los desarrollos que le suceden, precisamente de la mano de las contribuciones singulares de nuestro protagonista.
La carrera por el Genoma Humano
Dos años antes del inicio del PGH, los Institutos Nacionales de la Salud (NIH) de EEUU crearon la Oficina para la Investigación del Genoma Humano, posteriormente denominado Centro Nacional para la Investigación del Genoma Humano y finalmente Instituto Nacional para la Investigación del Genoma Humano (NHGRI). Bajo la forma de Consorcio implicó a centenares de científicos investigando simultáneamente en 20 centros de secuenciación en Alemania, China, Francia, Gran Bretaña, Japón y los Estados Unidos. Para una mayor efectividad se anunció un plan a 15 años (1990-2005) con los siguientes objetivos:
a) Determinar la secuencia completa de los varios miles de millones de nucleótidos del DNA humano y localizar los genes estimados entonces entre 50.000 y 100.000.
b) Construcción de mapas físicos y genéticos.
c) Analizar genomas de organismos usados como sistemas modelo en investigación.
d) Estudiar y debatir las implicaciones éticas y legales tanto para los individuos como para la sociedad.
En paralelo con los objetivos señalados, se desarrollaron métodos informáticos rápidos y sofisticados que permitieron acelerar el proceso de secuenciación y cartografiado del genoma. Pero antes de entrar en materia, vamos a comentar los antecedentes de este singular proyecto.
En 1984, el biólogo molecular Robert Sinsheimer, rector de la Universidad de California en Santa Cruz, propuso la creación de un Instituto para secuenciar el Genoma Humano. Desafortunadamente, Sinsheimer no recibió la financiación para dicho proyecto. Independientemente de los esfuerzos de Sinsheimer, el Departamento de Energía (DOE) de EE.UU. trabajaba en esta área. Durante la posguerra, el DOE había mostrado interés por la genética humana con la finalidad de comprender los efectos de la radiación en el material genético. Una de las principales técnicas empleadas en este trabajo fue el examen visual de los cromosomas en busca de anormalidades inducidas por la radiación. En 1983, los dos principales laboratorios de armamento nuclear (Los Álamos y el Lawrence Livermore), ya trabajaban en un proyecto denominado Biblioteca Génica. En 1986 se habían obtenido mapas y bibliotecas de genes de todos los cromosomas, salvo el 10 y el 11. En febrero de 1986 los laboratorios nacionales habían conseguido elaborar una «biblioteca» de fragmentos de DNA humano.
El Departamento de Energía, DOE, cuenta con una Oficina de Investigación Sanitaria y Ambiental (OHER), encargada de supervisar la seguridad en los trabajos con radiaciones. En 1986, Charles DeLisi, director de la OHER, propuso que el DOE aumentara su participación en las investigaciones genéticas basadas en la nueva biología molecular. Era consciente de que la secuenciación del genoma humano sería una tarea ardua pero aseguraba que el DOE, con sus dos grandes laboratorios nucleares, podría asumir dicha responsabilidad. En ese año la comunidad científica asumía que el mapeo y la secuenciación del genoma humano representaban un gran avance para las ciencias de la vida, pero varios de sus miembros eran conscientes de las discrepancias acerca de cuál era el mejor camino para alcanzar el objetivo. En un editorial de la revista Science, el director , Daniel Koshland expuso de un modo claro y conciso los argumentos a favor del Proyecto Genoma: «La principal razón por la que el Congreso apoya la investigación en otras especies es que puede resultar aplicable a los seres humanos. Por lo tanto, la respuesta obvia a la pregunta de si se debe descifrar la secuencia del genoma humano es Sí. ¿Por qué lo pregunta?»
El 1 de octubre de 1988, James Watson, uno de los descubridores de la estructura del DNA, fue nombrado Director Asociado de la Investigación del Genoma Humano en el NIH, con un presupuesto de más de 28,2 millones de dólares para el período 1988-1989 (unos 10 millones más que el presupuesto del DOE para investigar el genoma el mismo año). Aquel mismo día, el NIH y el DOE firmaron un Memorándum de Entendimiento, en el que las dos agencias se comprometían a cooperar en la investigación del genoma. El Proyecto Genoma Humano de EE.UU. había comenzado la carrera y liderado por el NIH. James Watson dimitió por diferencias acerca de cómo dirigir el proyecto genoma humano y fue relevado por Francis Collins.
Ante el creciente interés internacional por el Proyecto Genoma, se hizo necesaria la existencia de un foro internacional. En 1988, durante una reunión celebrada en Cold Spring Harbor, los investigadores fundaron la Organización del Genoma Humano (HuGO), cuyo cometido sería coordinar los trabajos internacionales con el fin de evitar solapamientos y repeticiones. La sede oficial de HuGO está en Ginebra, pero sus oficinas operativas se hallan repartidas en Londres, Bethesda y Osaka. Aunque empezó a funcionar con fondos aportados por organizaciones benéficas, como la británica Wellcome Foundation Trust, la HuGO al carecer de fondos propios para financiar la investigación, se encontró con dificultades de aceptación como interlocutor. Por otra parte, la UNESCO declaró el Genoma Humano Patrimonio Universal del Humanidad, evitando así que las empresas y agencias involucradas pudieran apropiarse del nuevo conocimiento y que una nueva barrera se sumara a las ya existentes entre los países avanzados y los más rezagados en materia económica y tecnológica.
Durante los años noventa se realizó un avance espectacular en la identificación de genes causantes de enfermedades pero Collins sabía que estos éxitos no servían como justificación por sí solos para la gran inversión económica realizada en el Proyecto Genoma. Collins desde el preciso momento en que se responsabilizó del proyecto, asumió que hacían falta dos cosas para poder finalizar la secuenciación del genoma humano, prevista para el año 2012. Lo primero era elaborar un mapa físico completo de cada cromosoma, formado por una serie de fragmentos de DNA. La segunda era conseguir mejoras relevantes en la eficacia y velocidad del proceso de secuenciación.

En 1994, Craig Venter, licenciado en Bioquímica y doctor en fisiología y farmacología, quien hasta ese momento dirigía las investigaciones en uno de los centros del NIH, creó con financiación mixta, el Instituto para Investigación Genómica (TIGR) y dirigió el desciframiento de la secuencia completa del genoma de la bacteria H. influenzae. En mayo de 1998 fundó su propia compañía Celera Genomics, resultante de la unión de Applera Corp. y TIGR. Celera firmó un acuerdo comercial de inversión conjunta a largo plazo con Applied Biosytems para la comercialización de los resultados de sus hallazgos. En enero del año 2000 Celera Genomics sorprende a la comunidad científica al anunciar que ha secuenciado el 90% del genoma humano.
En el verano, Craig Venter, presidente de Celera Genomics, y Francis Collins, director del Proyecto Genoma Humano, realizan un primer anuncio conjunto manifestando que han completado la secuenciación del genoma humano. Los borradores del genoma humano fueron presentados el 26 de junio en la Casa Blanca. En febrero de 2001 Celera publicó la secuenciación del genoma en la revista ‘Science’. El consorcio público hace lo mismo en ‘Nature’. Este momento marca la consolidación de la Era de la Genómica y el comienzo de una nueva Era, la Post-genómica.


Creación de Celera Genomics.
Celera fue fundada en 1998 con la misión secuenciar el genoma humano y ofrecer a los clientes un pronto acceso a los datos resultantes. Mediante el uso de la tecnología de secuenciación suministrada por Applied Biosystems y una sofisticada informática desarrollada a nivel interno, Celera fue pionero en la aplicación de la técnica de secuenciación denominada «shot gun”. Si bien esta técnica fue un enfoque muy criticado en su momento, se convirtió posteriormente en un método estándar para la secuenciación de organismos complejos que ahora es ampliamente aceptado y utilizado habitualmente por muchos aquellos científicos que originalmente despreciaron este método. Celera se embarcó en la construcción de un negocio de bases de datos, proporcionando herramientas personalizadas de búsqueda y software que permitió a decenas de compañías farmacéuticas y cientos de clientes académicos, gubernamentales y de biotecnología utilizar sus resultados en la investigación biológica. Mientras que el negocio de bases de datos de Celera en última instancia se convirtió en rentable, estaba claro en el año 2000 que no era un modelo de negocio sostenible por el libre acceso a las secuencias del genoma.

La empresa creada por Craig Venter, avanzó tanto científica como comercialmente. Científicamente, se reconoce que la comprensión de las enfermedades complejas como el Alzheimer y la enfermedad cardiovascular requiere una mayor comprensión de la variabilidad genética humana y por lo tanto, se embarcó en la iniciativa de Applera Genomics. Se trataba de volver a secuenciar los genes y regiones reguladoras de 39 personas para desarrollar un catálogo de la variabilidad genética humana. Se centraron en aquellas partes del genoma que codifican proteínas y por lo tanto, tenían mayor probabilidad de impacto en la salud humana. Este esfuerzo se tradujo en la identificación de más de 40.000 nuevos SNPs funcionales (polimorfismos de nucleótido único). Este trabajo se ha convertido en la base para muchas de las nuevas pruebas genéticas que Celera está desarrollando.
En enero de 2006, Celera anunció su intención de asociarse a programas de fármacos de moléculas pequeñas y adquirió los derechos para Celera Diagnostics, que había sido antes conjuntamente con Applied Biosystems.
En la actualidad, Celera es una compañía sanitaria centrada en la personalización de la gestión de la enfermedad a través de productos y servicios de diagnóstico.
Avances en la secuenciación automatizada.
La historia de Venter y Celara se remonta a 1986 cuando Venter viajó a California para entrevistarse con Michael Hunkapiller, uno de los diseñadores de una nueva máquina para la secuenciación automática de DNA fabricada por Applied Biosystems (ABI), una división de Perkin Elmer Corporation (PE). Junto con Leroy Hood, profesor de biología en Caltech, había desarrollado un método avanzado para secuenciación del DNA. La técnica seguía empleando el método de interrupción de la cadena ideado por Sanger pero este nuevo sistema empleaba tintes fluorescentes en lugar de radiactivos [2].
Un cebador para la secuenciación se marcaba fluorescentemente con cuatro colores distintos. Cada cebador marcado distinto era empleado en cada una de las cuatro reacciones de secuenciación dideoxi. Estas reacciones se combinaban y se sometían a electroforesis en gel de poliacrilamida en tubo. El DNA era observado por fluorescencia al pasar un detector en la parte inferior del gel y los cuatro colorantes se distinguen por sus colores. Los datos de fluorescencia se registran de forma continua y almacenados por un equipo. ABI desarrolló programas para interpretar automáticamente los datos para producir una secuencia real. La secuencia podía ser deducida por el orden en que los cuatro colores pasaban por el detector. Esta técnica permitía prescindir de la radiactividad. Además, puesto que cada letra de DNA iba marcada con un tinte fluorescente distinto, la mezcla de cadenas de DNA de una reacción de secuenciación se podía hacer pasar por una vía del gel, aumentando así su capacidad de secuenciación. El ABI 373A podía analizar 24 muestras de forma simultánea con un rendimiento de 12000 letras (nucleótidos o bases) al día.
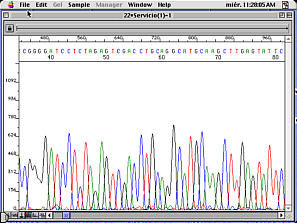
El secuenciador de DNA ABI 370A fue utilizado por primera vez para determinar la secuencia de un gen por Craig Venter en el NIH . En 1987 el laboratorio de Venter en el NIH se estableció un servicio de secuenciación con secuenciadores ABI y del prototipo de terminal robotizada ABI 800, que mezclaba DNA, productos químicos y enzimas para las reacciones de secuenciación. Gracias al ABI373A, Ventor y sus colaboradores secuenciaron cerca de 60000 bases de DNA en las inmediaciones del gen de Huntington y unas 106000 de la región candidata para la distrofia muscular.
Una demostración de la potencia de la secuenciación automatizada fue el desarrollo de las etiquetas de secuencia expresada (EST), que eran los fragmentos de cada secuencia de cDNA. En este enfoque, copias cDNA del RNA mensajero se clonaban al azar y eran sometidas a la secuenciación automatizada. En el primer informe de Venter en 1991, se descubrieron 337 nuevos genes humanos, 48 de ellos homólogos a genes de otros organismos.
Este estudio inicial se amplió con un análisis de 83 millones de nucleótidos de la secuencia de cDNA que identificaron fragmentos de más de 87 000 secuencias de cDNA humano, más de 77 000 de los cuales eran desconocidos. Este enfoque fue adoptado por muchos de los proyectos genoma. Hoy en día la base de datos de EST contiene más de 43 millones de ESTs de más de 1300 organismos distintos. Otra aplicación temprana del secuenciador automático fue el proyecto de secuenciación del genoma del gusano, que estaba en marcha en 1992.
El Instituto de Investigación Genómica (TIGR) fue creado por Venter en 1992, con el objeto de expandir su operación de secuenciación. Instaló 30 secuenciadores automáticos ABI 373A, diecisiete estaciones ABI Catalyst y una base de datos instalada en un sistema Sun SPARCenter2000. Esta era una fábrica real con equipos dedicados a distintas etapas del proceso de secuenciación como la preparación del molde, el gel de vertido y funcionamiento del secuenciador. El análisis de los datos se integraba en el proceso para que los problemas en las etapas tempranas puedan ser detectados y corregidos tan pronto como sea posible. Sus objetivos eran dilucidar la posición de cada gen en cada uno de los 23 pares de cromosomas, multiplicar por diez la producción de EST y comenzar la secuenciación de varios de los genes de los organismos modelo.
En 1993, el Centro Sanger, posteriormente conocido como el Wellcome Trust Sanger Institute, fue establecido por el Wellcome Trust y el Medical Research Council. Este es posiblemente el más importante de los centros de secuenciación establecidos para participar en el esfuerzo mundial para la secuenciación el genoma humano. Situado en Hinxton, cerca de Cambridge, «el Sanger» es un ejemplo sobresaliente de un centro de secuenciación moderna. La instalación ha producido 3.4×109 bases de la secuencia terminada por el 30 aniversario del método de secuenciación didesoxi.
Una de las contribuciones del equipo de Venter en los primeros pasos en la carrera para la secuenciación del genoma humano, fue básicamente metodológica: la secuenciación “shotgun”. Este descubrimiento fue en gran medida responsable del adelanto en el plazo de terminación del primer borrador del genoma humano. El “shotgun” es una técnica de laboratorio para determinar la secuencia de DNA del genoma de un organismo. El método consiste en romper el genoma en una colección de pequeños fragmentos de DNA que pueden ser leídos por las máquinas de secuenciación automatizada. A continuación, con programas especializados de software, se buscan coincidencias en las secuencias de DNA y se procede a colocar los fragmentos individuales en el orden correcto para reconstruir el genoma. Debido a las grandes cantidades de datos que se generan y a la complejidad de los genomas de los organismos, los programas de éxito se basan en algoritmos complejos, basados en los conocimientos de campos tan diversos como la estadística, la teoría de grafos, la informática y la ingeniería informática.
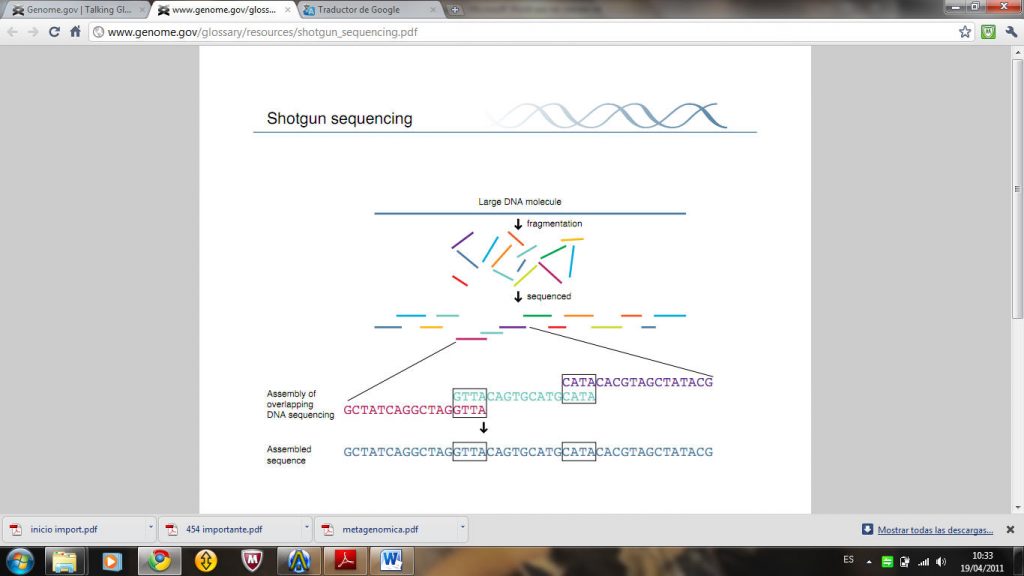
Esta técnica intenta superar las limitaciones de las tecnologías de secuenciación al romper el DNA al azar. Las técnicas de secuenciación sólo eran capaces de «leer» varios cientos de nucleótidos a la vez. Las piezas resultantes se juntaron basándose en la similitud entre las piezas procedentes de la misma sección de la molécula de DNA original .La gran cantidad de datos producidos hizo necesario el empleo de programas de ordenador para realizar el ensamblaje. A pesar de las continuas mejoras en la tecnología de secuenciación y el desarrollo de especializados programas de ensamblaje, no estaba claro si la secuenciación shotgun podría ser utilizado para secuenciar genomas más grandes que los de los virus (por lo general 5000-100,000 nucleótidos). Para grandes genomas se pensó que la complejidad de la tarea representaría un desafío insuperable para cualquier programa de ordenador.
En 1995, sin embargo, los investigadores del Instituto de Investigación Genómica (TIGR) utilizaron con éxito la técnica del “shotgun” para descifrar el genoma completo de la bacteria Haemophilus influenzae. La secuenciación de 1,83 millones de pares de bases exigió el desarrollo de un programa especializado de ensamblaje así como denodados esfuerzos para completar aquellas regiones que no podían ser correctamente ensambladas por el software. El éxito del proyecto Haemophilus inició una revolución de la genómica con el número de genomas secuenciados que cada año crece a un ritmo exponencial. Hasta el momento los genomas de más de 1000 virus, 100 bacterias, y varios eucariotas han sido completados, mientras que otros tantos proyectos están camino a su finalización.
La revolución genómica llevó al nacimiento de un nuevo campo: la Bioinformática, que reúne una mezcla ecléctica de los campos científicos, tales como ciencias de la computación y la ingeniería, las matemáticas, la física, la química y la biología.
Los detractores de la secuenciación “shotgun” siguieron cuestionando su aplicabilidad a los genomas grandes a pesar de los éxitos evidentes en la secuenciación de genomas bacterianos. Ellos argumentaban que la técnica sería impracticable en el caso de grandes genomas eucariotas porque regiones de DNA que se producen en dos o más copias en el genoma conducirán a error a cualquier ensamblador. El procedimiento estándar para el manejo de grandes genomas consiste en romper el DNA en grandes piezas (50 a 150 kpb) ,seguido del clonado en cromosomas artificiales bacterianos (BAC),y, a continuación secuencia cada BAC a través del método de “shotgun”.
La mayoría de esas críticas se silenciaron en el año 2000 por el ensamblaje del genoma de Drosophila melanogaster realizado por Celera. El ensamblaje se realizó con un nuevo ensamblador diseñado para manejar los problemas específicos que surgen en el ensamblaje de grandes genomas complejos. Los investigadores de Celera procedieron a ensamblar el genoma humano utilizando la técnica de secuenciación “shotgun” del genoma completo (WGS) [4]. Sus resultados fueron publicados simultáneamente con las del Organismo Internacional de Secuenciación del Genoma Humano del Consorcio, que utilizaron el método jerárquico tradicional. Estudios independientes posteriores mostraron que los dos conjuntos de resultados similares, y muchas de las diferencias entre ellos podría ser explicado por la calidad del proyecto a nivel de los datos.
El éxito de Celera, combinada con las ventajas de costo de la técnica del WGS y la secuenciación y ensamblado el genoma humano en un poco más de un año, mientras que el Consorcio Internacional llevaba embarcado en dicha tarea desde hace más de 10 años-
Creó un gran interés en el método WGS y llevó al desarrollo de varios programas de ensamblaje WGS : Aracne en el Instituto Whitehead, Phusion en el Instituto Sanger, Atlas en el Centro de Secuenciación del Genoma Humano Baylor, y el Jazz en el DOE Joint Genome Institute. La mayoría de los proyectos de secuenciación actuales han optado por un enfoque WGS en lugar del enfoque tradicional. Por ejemplo, la secuenciación del ratón, la rata, perro, pez globo, y ascidia todos siguen la estrategia de WGS.

Biología sintética.
El tiempo empleado en terminar la tarea de descifrar genomas completos es reflejo del grado de complejidad y de la evolución de la genómica desde sus comienzos. El primer genoma completo fue el de un virus. En 1977, Sanger y sus colegas determinaron la secuencia genética completa del fago φX174. Pero hubo que esperar dieciocho años, en 1995, para terminar la primera secuencia genética completa de una bacteria, Haemophilus influenzae [5].
La lectura de la secuencia genética de una amplia gama de especies ha aumentado de manera exponencial a partir de estos primeros estudios. La capacidad de digitalizar con rapidez la información genómica se ha incrementado en más de ocho órdenes de magnitud en los últimos 25 años.
¿Se pueden explicar todas las funciones celulares a partir de su genoma? ¿Contiene el genoma la información necesaria para el funcionamiento de una célula? ¿Se puede sintetizar un genoma artificialmente y que sea tan funcional como el original? Estas preguntas dieron lugar a una iniciativa de investigación conocida como Biología Sintética. Para ello Craig Venter creo la compañía Synthetic Genomics Inc. Agrupando un esfuerzo inversor notable en los Estados Unidos (figura).
El interés en la síntesis de moléculas de DNA de gran tamaño y los cromosomas surgió a partir de los esfuerzos del grupo de Venter en los últimos 15 años para construir una célula mínima que contuviese sólo los genes esenciales. La idea consistía en ir eliminando genes de una bacteria hasta determinar los genes imprescindibles, es decir, el conjunto mínimo de genes que puede tener esa bacteria para funcionar en un ambiente rico en nutrientes. Este trabajo fue finalizado en 1995, cuando la secuencia del genoma de Mycoplasma genitalium, una bacteria con el mínimo genoma conocido capaz de un crecimiento independiente en el laboratorio. Más de 100 de los 485 genes que codifican proteínas de M. genitalium son prescindibles.
El siguiente reto fue desarrollar una estrategia para el ensamblaje de fragmentos de tamaño viral para producir una molécula completa de DNA. Con este objetivo se planteó reunir la síntesis del genoma de M. genitalium en cuatro etapas, desde “cintas” de DNA sintetizado con un promedio de 6 KB de tamaño. Esto se logró mediante una combinación de métodos enzimáticos in vitro y por recombinación en Saccharomyces cerevisiae in vivo. Una vez finalizado, todo el genoma sintético [582.970 pares de bases] fue introducido y replicado de manera estable como un plásmido dentro de levadura.
Superados diversos obstáculos para la introducción y expresión de un cromosoma de síntesis química en una célula receptora, se combinaron todos diversos procedimientos y se procedió a la síntesis, ensamblaje, clonación e introducción del genoma de 1,08 Mbp de M.mycoides en células de levadura para crear una nueva célula controlada por este genoma.
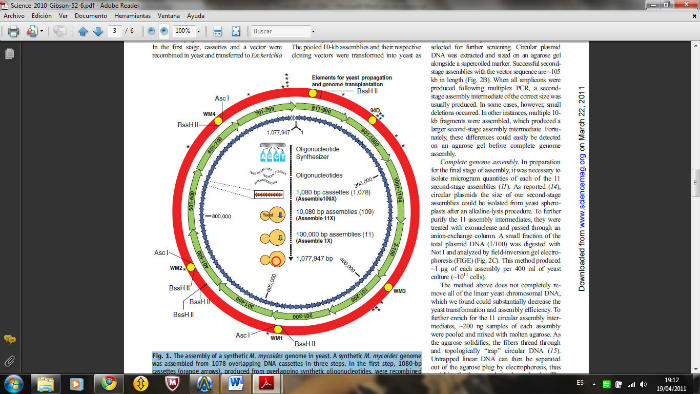
El siguiente reto afrontado por el grupo de Venter consistió en sintetizar de novo el genoma completo de una bacteria, M. Mycoides, es decir unir uno a uno en su correcto orden todos los nucletidos que forman la molécula de DNA. En 1995, se admitía un error de 10.000 pares de bases, y la secuenciación de un genoma microbiano podía requerir meses. Hoy en día, la precisión es mucho mayor y la secuenciación sólo requiere unos pocos días. No obstante, crear un genoma sintético libre de errores para crear un “nueva” célula controlada únicamente por dicho genoma sintético es complicado y requiere un estricto control. La demostración de que un genoma sintético da lugar a células con las características de las células M. mycoides implica que la secuencia de DNA en la que se basa es lo suficientemente precisa para especificar una célula viva con las propiedades adecuadas. Este enfoque de la genómica sintética se encuentra en profundo contraste con otros enfoques distintos de ingeniería que modifican los genomas naturales mediante la introducción de múltiples inserciones, sustituciones o deleciones. Este trabajo ofrece una prueba inicial para la producción de células sobre la base de diseño por ordenador de secuencias del genoma. La secuenciación del DNA de un genoma permite el almacenamiento de las instrucciones genéticas para la vida como un archivo digital. El genoma sintético creado tiene limitadas variaciones respecto del genoma natural de M. mycoides. Sin embargo, el enfoque desarrollado por Venter debe ser aplicable a la síntesis y trasplante de genomas más novedosos. En sus trabajos, Venter se refiere a una célula controlada por un genoma ensamblado a partir de fragmentos de DNA obtenidos por síntesis química como una «célula sintética,» a pesar de que el citosol y la membrana de la célula receptora no son sintéticos. Los efectos fenotípicos del citoplasma receptor se diluyen con el recambio de las proteínas. Después del trasplante y la reproducción en una placa para formar una colonia (> 30 divisiones o dilución de 109 veces), la progenie no contendrá ninguna moléculas de proteína que estaban presentes en la célula receptora original. Las propiedades de las células controladas por el genoma ensamblado se espera que sea el mismo que si toda la célula se hubiera producido de manera sintética (el software de DNA construye su propio hardware).
Si los métodos descritos aquí se pueden generalizar, el diseño, la síntesis, el ensamblaje, y el trasplante de cromosomas sintéticos ya no será un obstáculo para el progreso de la biología sintética. Se espera que los costes en la síntesis de DNA desciendan tal como ocurrió con la secuenciación de DNA. Un menor coste de síntesis junto con la automatización, permitirá amplias aplicaciones de la genómica sintética.
ectos de investigación, particularmente en el campo emergente de la biología sintética.
Metagenómica
Los avances en la capacidad de secuenciación de genomas acontecidos en los últimos años ha dado lugar a un nuevo campo de la ciencia denominado “metagenómica”. Este emergente campo busca examinar el contenido genómico de las comunidades de organismos para entender sus funciones e interacciones en un ecosistema. Dado el papel de gran alcance que los microorganismos desempeñan en muchos ecosistemas, los estudios de metagenómica de las comunidades microbianas pueden ofrecer una visión de las familias de proteínas y su evolución. Debido a que la mayoría de los microorganismos objeto de estudio no crecen en el laboratorio utilizando las técnicas habituales de cultivo, los científicos han recurrido a técnicas independientes de cultivo para estudiar la diversidad microbiana. El método de secuenciación “shotgun” permite un muestreo aleatorio de secuencias de DNA para examinar el material genómico de una comunidad microbiana. El proyecto bandera de este nuevo campo consistió en la secuenciación “shotgun” para examinar las comunidades microbianas en muestras de agua recogidas en la expedición Sorcerer II [8], también dirigido por nuestro conocido Craig Venter. Este análisis predijo más de seis millones de proteínas en datos GOS, casi el doble de la cantidad de proteínas presentes en las bases de datos en ese momento. Estas predicciones añaden gran diversidad a las familias de proteínas conocidas y abarcan casi todas las familias conocidas de proteínas procariotas. Algunas de las proteínas encontradas no tenían similitud con las proteínas conocidas actualmente, por lo que representan nuevas familias. Una mayor fracción esperada de estas nuevas familias se prevé que sea de origen viral. También se encontró que varios dominios de proteínas que se pensaba hasta entonces que eran específicos de reino, se han encontrado GOS en otros reinos. Este procedimiento abre la puerta a una multitud de análisis de familias de proteínas e indica que estamos muy lejos del muestreo de todas las familias de proteínas que existen en la naturaleza.
Pero la metagenómica no solo se ha ocupado del estudio de comunidades bacterianas en el medio natural. Recientemente se han inciado proyectos en diversos laboratorios para estudiar lo que se denomina el microbioma humano, es decir el conjunto de microorganismos que conviven de modo simbiótico en el cuerpo humano. Un ejemplo claro es la flora intestinal, compuesta por cientos de especies de microorganismos, principalmente bacterias.
Medicina Personalizada
Ademas de ayudar a la identificación de posibles enfermedades de origen genético, el mapa del DNA específico de una persona permite comprender a qué medicamentos responde mejor esta persona. Esto ha dado lugar al desarrollo de la medicina personalizada.
Los padres de los estudios sobre el genoma, Craig Venter y James Watson compararon sus mapas genéticos para mostrar cómo puede la medicina personalizada evitar efectos indeseables de los fármacos y encontrar genes que predispongan a un individuo a parecer una determinada enfermedad. El primer mapa genético humano se basó en el genoma de Venter, mientras que el de Watson fue analizado en 2007 y ahora los dos son públicamente accesibles. Craig Venter declaró en The New York Times, que es portador de la variante genética apoE4, relacionada con deficiencias en el metabolismo de lípidos, y el riesgo de padecer Alzheimer.
Existe un gran interés en la caracterización de la variación en un humano individual, ya que esto puede aclarar lo que contribuye de manera significativa con el fenotipo de una persona, lo que permite la genómica personalizada. Venter y sus colaboradores investigaron las variantes en el «exoma» de una persona, que es el conjunto de exones en el genoma, ya que el exoma se cree que alberga gran parte de la variación funcional que diferencian al individuo [10].

En resumen, somos protagonistas, queramos o no, de una revolución que afecta ya a nuestras vidas. Este camino emprendido por la genómica ha supuesto una vertiginosa carrera de manos de unas tecnologías que van por delante de nuestra capacidad de análisis, hecho insólito en las ciencias de la Vida. Esta circunstancia ha situado la Bioinformática como protagonista indispensable para poder analizar los resultados experimentales.
La metagenómica nos revela la gran biodiversidad que nos rodea, pero este complejo mundo exterior también se refleja en el interior del individuo: el “bioma” humano. La presencia de infinidad de microorganismos en autentica simbiosis con el cuerpo humano demuestra que como individuos no somos una sola especie, si no que nuestra existencia depende de una colección de otras especies sin las cuales no podemos sobrevivir. El microbioma humano se considera ya un nuevo órgano del cuerpo, como el corazón, el hígado o el sistema nervioso.
El ser humano desde que nace está expuesto a un gran número de agentes externos que interaccionan con él y requieren una respuesta adecuada. El “exposoma”, como así se denomina esta relación con el ambiente, determina enfermedades y configura el propio sistema inmune. En definitiva el ser humano esta configurado por un genoma, innumerables genomas de su bioma y los agentes incluidos en el exposoma. El propio concepto de ser humano supera al individuo y lo sitúa en otra dimensión, insospechada ni siquiera hace una década.
Bibliografía
[1] Collins, F, Morgan, M., Patrinos, A. The Human Genome Project: Lessons from Large-Scale Biology. Science: 286 Apr 11 (2003).
[2] DNA sequencing: bench to bedside and beyond Clyde A. Hutchison III.J. Craig Venter Institute, 9704 Medical Center Drive, Rockville, MD 20850, USA.2007.
[3] Shotgun Sequence Assembly. MIHAI POP. The Institute for Genomic Research (TIGR) Rockville, USA.
[4] Whole-genome shotgun assembly and comparison of human genome assemblie. Sorin Istrail, Granger G. Sutton,a Liliana Florea, Craig Venter et al. Proc Natl Acad Sci U S A.PNAs. February 17; 2004.101(7): 1916–1921.
[5] Creation of a bacterial cell controlled by a chemically synthesized genome.
Gibson DG, Glass JI, Lartigue C,Venter JC et al.Science. 2010 Jul 2;329 (5987):52-6. Epub 2010 May 20.
[6] One-step assembly in yeast of 25 overlapping DNA fragments to form a complete synthetic Mycoplasma genitalium genome.Daniel G. Gibson. Craig Venter, Hamilton, O. Smith et al. The J. Craig Venter Institute, Synthetic Biology.PNAs.2008
[7]. Holt RA, Warren R, Flibotte S, Missirlis PI, Smailus DE (2007) Rebuilding microbial
genomes. Bioessays 29:580–590.
[8]The Sorcerer II Global Ocean Sampling Expedition: metagenomic characterization of viruses within aquatic microbial samples. Williamson SJ, Rusch DB, Yooseph S, Venter JC et al. PLoS One. 2008 Jan 23;3(1):e1456.
[9] Structural and functional diversity of the microbial kinome. Kannan N,Taylor SS,Zhai Y,Venter JC,Manning G. PLoS Biol. 2007;5(3):e17.
[10] Genetic Variation in an Individual Human Exome. Pauline C. Ng*, Samuel Levy, Jiaqi Huang, Timothy B. Stockwell, Brian P. Walenz, Kelvin Li, NelsonAxelrod, Dana A. Busam, Robert L. Strausberg, J. Craig Venter. PLoS Genetics. 2008. 4 (8)
Alba de Lorenzo Avilés y Federico Morán

Federico Morán
Catedrático de Bioquímica de la Universidad Complutense de Madrid
SIMBIONTE
Especies simbióticas
Conforman mi Bioma
Cromosomas y Exposoma Mi auténtico Metagenoma
¿Quién soy? ¿Qué soy?
¿Qué somos? Simbiontes
¿Quién soy? ¿Qué soy?
¿Qué somos? Simbiontes
Genómica, Transcriptómica
Postgenómica, Metagenómica
Fluxómica, Metabolómica
Postgenómica, Metagenómica
Genoma, cromosoma
Bioma, Exposoma
Simbiontes, simbiontes
Somos simbiontes
Genómica, Transcriptómica
Postgenómica, Metagenómica
Fluxómica, Metabolómica
Postgenómica, Metagenómica
Genoma, cromosoma
Bioma, Exposoma
Simbiontes, simbiontes
Somos simbiontes
AVIADOR DRO